.svg)

Evaluating Boltz-2 on Real Drug Targets: Does it work?
TLDR: Boltz-2 is a novel AI model for structure-based drug design that predicts both protein–ligand binding poses and binding affinities, reported to be faster than physics-based methods. In our early testing, it performs well on rigid, well-characterised proteins like KRAS and Mpro, accurately capturing binding interactions and structure–activity relationship (SAR) trends. However, it struggles with flexible, allosteric, or underrepresented targets (e.g., PI3K-α, WRN, cGAS), often missing ligand-induced conformational shifts or generating implausible poses. In some of these cases, its affinity predictions appear to be accurate. This highlights the need to determine whether Boltz-2’s affinity predictions are driven primarily by similarity to targets and ligands in its training set or by an authentic reconstruction of the underlying 3-D protein-ligand interactions. These early internal results show that, while promising, Boltz-2 works best on well-represented proteins and ligands and only with structural guidance or refinement on dynamic targets. A broad, rigorous, community-driven evaluation is needed to fully assess Boltz-2’s capabilities and domain of applicability.
Boltz-2 is a new AI model for structure-based drug design. In contrast to previous models like Boltz-1, Chai-1, and AlphaFold-3, it not only predicts protein structures but also the binding affinities between proteins and ligands. In other words, Boltz-2 can tell you both how a drug might fit into a protein and how well it might fit. And it’s fast – reported to be faster than traditional physics-based simulations for binding affinity. If Boltz-2 predictions are as accurate as physics-based methods, this speed will make Boltz-2 a versatile tool for structure-based drug discovery (SBDD), accelerating the hunt for new therapeutics.
To assess Boltz-2’s performance on real-world drug targets, we tested its co-folding accuracy on six protein–ligand systems. We evaluated two aspects: (a) whether it predicts realistic binding poses, and (b) whether its affinity predictions correlate with known SAR data. These six targets spanned two broad classes:
- Rigid globular proteins: These proteins maintain a relatively stable shape and are generally easier for modelling protein–ligand binding affinity.
- Flexible, multi-domain proteins: These proteins can rearrange their shape or only form the drug’s pocket when the ligand binds, making them far more challenging for any binding prediction.
For each protein in our test, we gave Boltz-2 the protein’s sequence (and the unbound structure, in some cases) and asked it to co-fold the protein with its ligand. We then analysed the model’s predicted complex and binding affinity for each case. When the model predicted multiple poses, we took an ensemble average of Boltz-2’s affinity predictions. It’s important to note that this was not an out-of-distribution study, and that we focused on publicly available protein–ligand complexes we were already familiar with, many of which may have appeared in Boltz-2’s training set. As such, strong performance on these examples, while promising, is not entirely surprising and should be viewed as an initial validation on well-represented targets rather than a test of the model’s generalisation capabilities.
Rigid - KRAS G12D (oncogenic KRAS mutant) + MRTX-1133
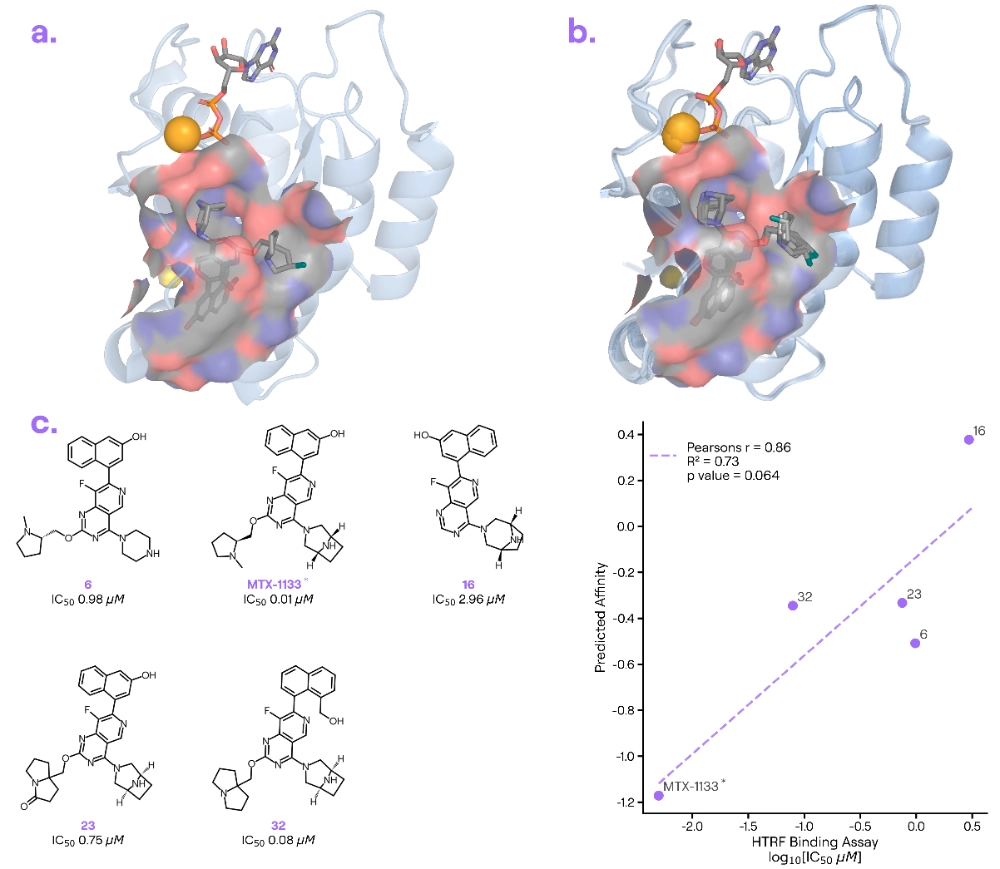
Target information: KRAS G12D is a notorious oncogenic mutation (found in colorectal and lung tumours) and a tough target due to subtle changes in its pocket.
Ligand information: MRTX-1133 [1] is a KRAS G12D inhibitor from Mirati Therapeutics (patented in 2021).
Binding poses: Boltz-2 captured the critical interaction between MRTX-1133 and the mutant D12 residue. The model even correctly modelled the closure of the protein’s Switch II loop—a key interaction that moulds the binding pocket for MRTX-1133 (Figure1 a, b)
Binding affinity: Boltz-2’s predicted binding affinities for MRTX-1133 and related analogues tracked known SAR trends. For example, modifications known to improve binding (e.g. adding groups that nestle into the lipophilic pocket near Switch II) were likewise predicted to increase affinity (Figure1 c, d)
Our take: The KRAS G12D–MRTX-1133 complex was first resolved in April 2022 (PDB: 7XKJ), and many related KRAS structures are also available. This case shows that for a well-structured target like KRAS, Boltz-2 can recapitulate important binding features and provide useful insights into what makes an inhibitor effective. (Note: The positive correlation seems driven by three clusters of points. Further, all five binding affinity measurements can be found in the ChEMBLv34 database, so it is highly likely that Boltz-2 was trained on these datapoints, ChEMBL assay ID CHEMBL5047302)
Rigid - SARS-CoV-2 Main Protease (Mpro) + Ensitrelvir
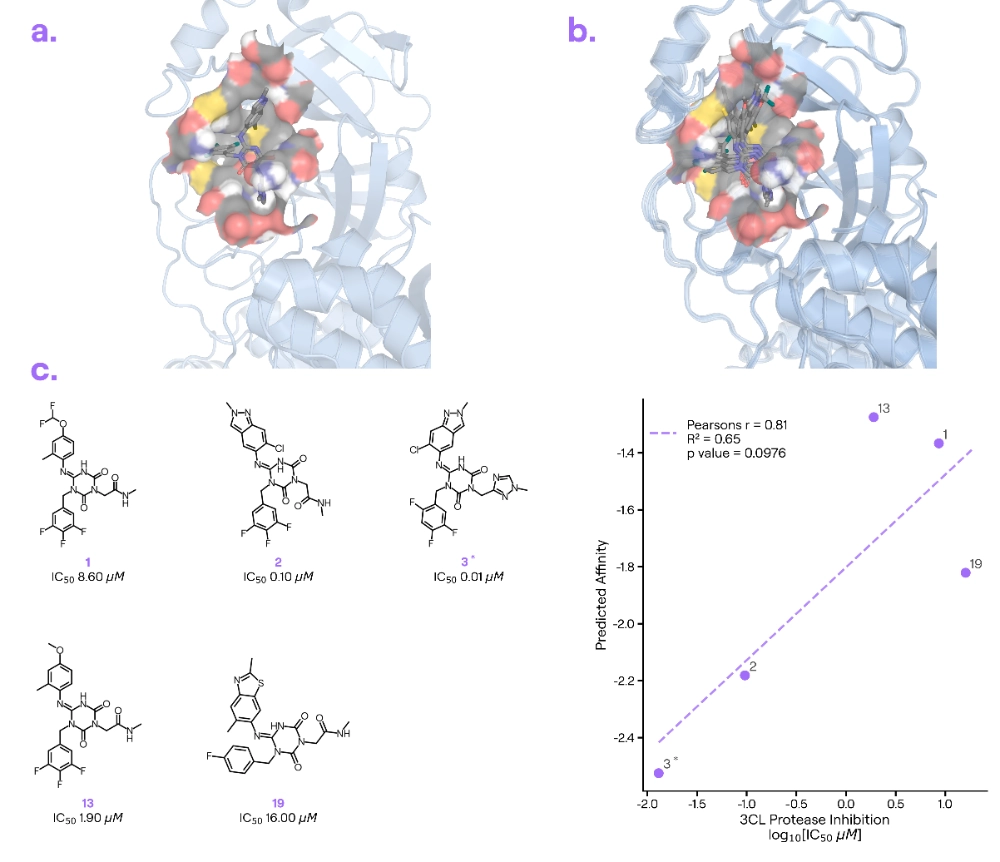
Target information: The coronavirus main protease (Mpro) is an antiviral target essential for viral replication,
Ligand information: Ensitrelvir [2] developed by Shionogi & Co., is a non-covalent inhibitor of Mpro.
Binding poses: Boltz-2 correctly identified Mpro’s substrate-binding pocket where Ensitrelvir binds. However, getting the exact pose proved tricky. Across multiple runs, Boltz-2 didn’t converge on a single consistent orientation for Ensitrelvir. In some predictions, it misplaced the molecule’s hydrophobic tail, and occasionally it even generated a chemically implausible ligand conformation (the kind that violates basic chemistry rules and would raise a medicinal chemist’s eyebrow).
Binding affinity: Despite the pose issues, Boltz-2’s affinity predictions for Ensitrelvir were in the right ballpark and even mirrored qualitative SAR trends.
Our take: Ensitrelvir’s Mpro complex was added to the PDB in June 2023 (PDB: 8HUR), and many similar Mpro structures were already known. These mixed pose results highlight the difficulty of sampling the ligand’s orientation. Surprisingly, Boltz-2’s affinity predictions remained accurate despite an imprecise pose. (Note: Also in this example, the good correlation in the binding affinity plot seems driven by three clusters of points. We found binding affinity values of molecules 1-3 in the ChEMBLv34 database, so it is highly likely that those were included in the training set, ChEMBL assay ID CHEMBL5163347.)
After seeing Boltz-2 perform on two rigid targets, we next challenged it with four flexible, multi-domain targets—cases that often require an induced fit and pose a tougher challenge for the model.
Flexible - PI3K-α (alpha isoform of PI3K kinase) + RLY-2608
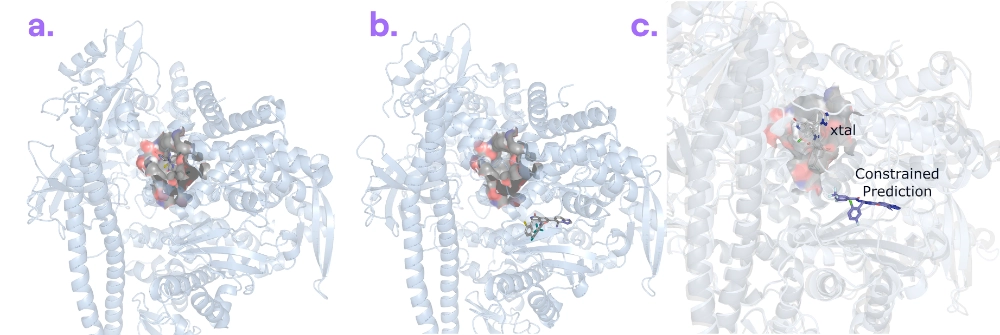
Target information: PI3K-α is a kinase enzyme in the PI3K/AKT signalling pathway and a high-priority target in oncology.
Ligand information: RLY-2608 [3] is a PI3K-α inhibitor from Relay Therapeutics.
Binding poses: Boltz-2 accurately predicted PI3K-α’s unbound structure, but it couldn’t reproduce the inhibitor-bound shape. The protein requires a subtle but important shift to accommodate RLY-2608, which Boltz-2 failed to capture. Without additional guidance, the model remained in PI3K-α’s default conformation and missed the alternate pocket shape required by the inhibitor.
Binding affinity: Not evaluated – the predicted pose was too far from reality to yield a meaningful affinity estimate.
Our take: This PI3K-α inhibitor complex (PDB: 8TSD) did not appear in the PDB until November 2023. The only other PDB entry with a ligand occupying the RLY-2608 pocket is STX-478 bound to PI3K-α (PDB: 8TDU), released in September 2023 - both after the Boltz-2 training cut-off. However, structures of PI3K-α with other inhibitors (binding in a different pocket) were available prior to that. This may have biased Boltz-2 and contributed to the incorrect prediction. Boltz-2’s failure to find the alternate conformation here underscores a limitation: if a protein doesn’t naturally adopt a certain shape, the model might not bend it into that shape just because a ligand is present. We even tried guiding Boltz-2 with pocket constraints toward the correct conformation; surprisingly, it still misplaced the molecule into an alternative pocket commonly used by other PI3K-α inhibitors, revealing a strong bias from the training set.
Flexible - WRN Helicase + HRO-761

Target information: WRN is a DNA helicase involved in DNA replication/repair, and is a promising cancer target (especially in certain microsatellite instability-high cancers where WRN is essential).
Ligand information: HRO-761 [4] is an allosteric inhibitor developed by Novartis that binds to WRN in a unique, ATP-independent conformation.
Binding poses: Boltz-2 readily predicted WRN’s known ATP-bound (apo) conformation. However, when we asked it to predict WRN bound to HRO-761 (an inhibitor that should push WRN into an alternate ATP-free conformation), the model kept reverting to the usual ATP-bound shape.
Binding affinity: Not evaluated – again, the predicted complex was incorrect, so any affinity prediction would be meaningless
Our take: The inhibitor-bound WRN structure (PDB: 8PFO) was deposited in April 2024, and no other WRN-inhibitor structures were available before mid-2023. Boltz-2 showed a strong bias toward WRN’s canonical shape (likely because the structure of ATP bound to WRN helicase was deposited on April 29, 2020 and most other training examples of WRN’s family, the RecQ helicases, were in the ATP-bound form). It struggled to escape that bias and find the alternate pose induced by HRO-761. This example highlights that Boltz-2 can overfit its training data and might n123eed explicit hints (e.g. templates or constraints) to explore less common conformations.
Flexible - cGAS + XL-3156
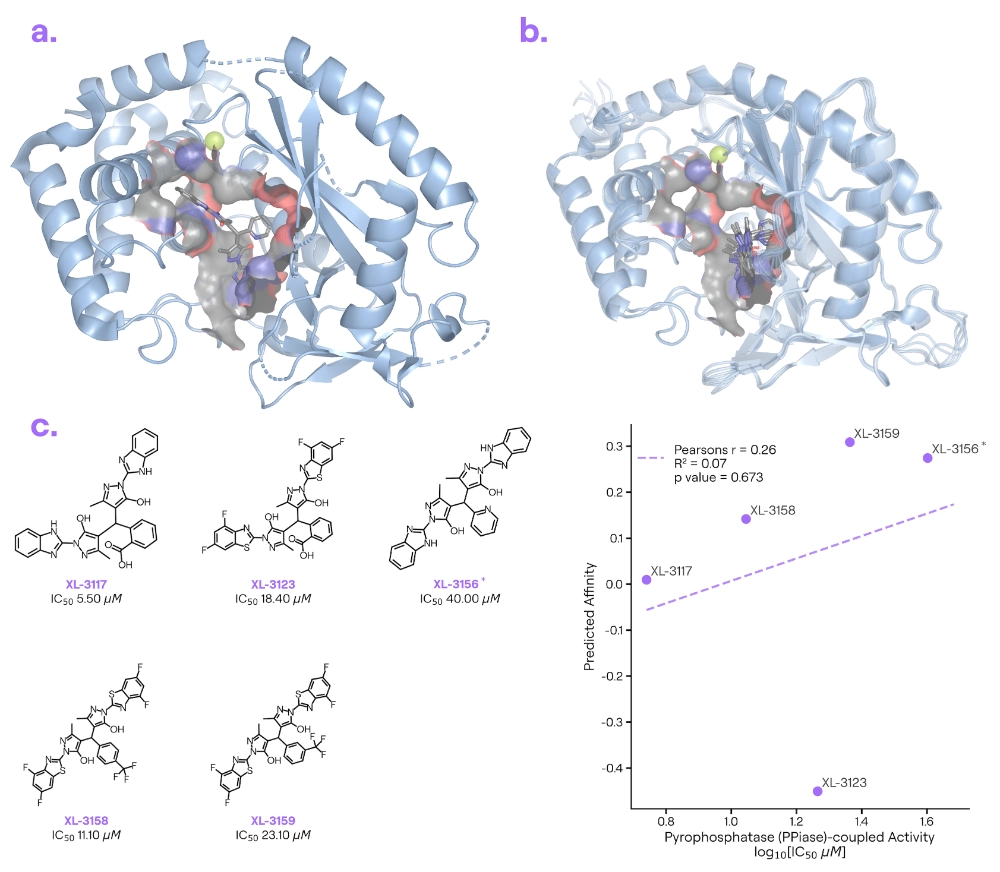
Target information: cGAS is an immune sensor that detects cytosolic DNA and triggers the cGAS–STING pathway (important in inflammation and autoimmune disease).
Ligand information: XL-3156 [5] is a small-molecule cGAS inhibitor from China Pharmaceutical University.
Binding poses: Boltz-2 correctly identified the general pocket on cGAS where XL-3156 binds, but it struggled with the ligand’s precise placement in that pocket.
Binding affinity: The compounds span a relatively narrow potency range, the weak positive correlation suggests no statistically significant relationship between predicted affinity and experimental values.
Our take: The inhibitor-bound cGAS structure (PDB: 9J2W) was deposited in June 2025, and other cGAS-inhibitor structures were available before June 2023. The predicted binding modes for XL-3156 were often chemically unrealistic (for example, Boltz-2 sometimes contorted the ligand’s bonds in ways actual chemistry wouldn’t allow). Likewise, the model’s affinity predictions for a series of XL-3156 analogues were mostly off. For a highly dynamic system like cGAS—where the protein may need to flex or subtle changes have big effects—Boltz-2’s current version has limited predictive power. It might get the pocket roughly right, but without capturing the induced fit, both the pose and the potency predictions will suffer.
Flexible - DHX9 + ATX-968
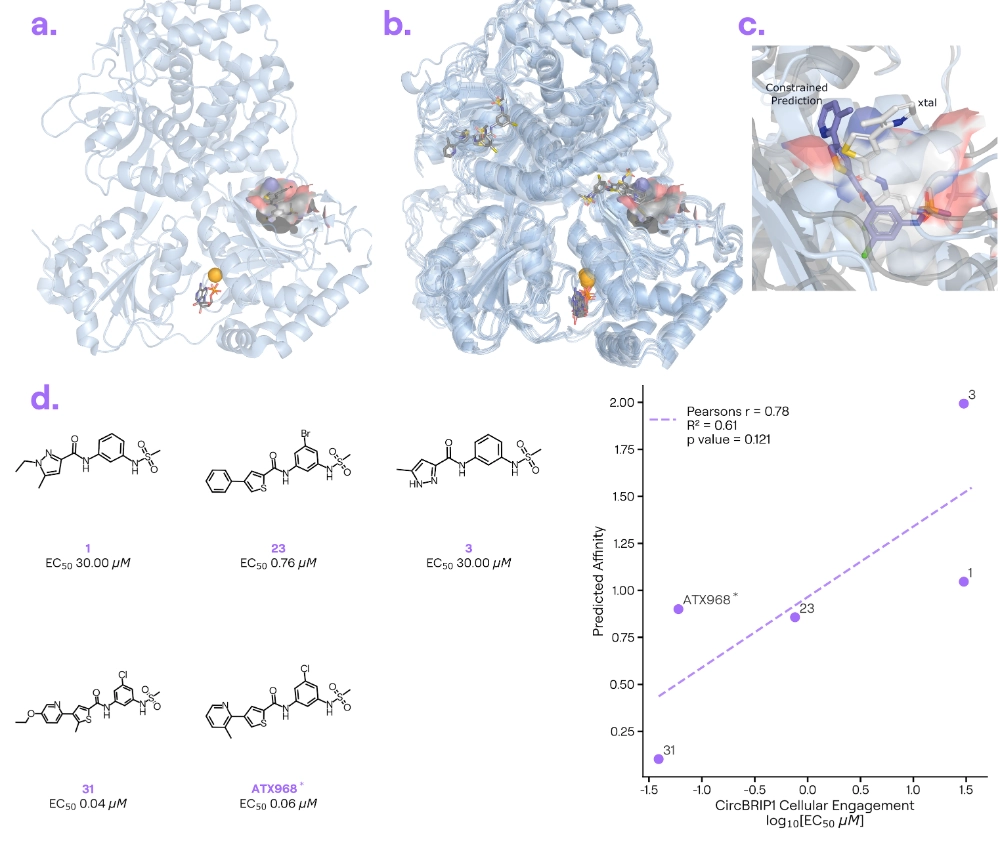
Target information: DHX9 is an RNA helicase involved in transcription regulation and genome stability, and it’s being explored as a cancer target. DHX9 has multiple domains, including an N-terminal region for which few structures are known.
Ligand information: ATX-968 [6] is a small-molecule inhibitor from Accent Therapeutics
Binding poses: Boltz-2 often misfolded DHX9’s N-terminal domain when attempting to co-fold the protein with ATX-968.
Binding affinity: Interestingly, Boltz-2’s predicted binding affinities for a series of ATX-968 analogues correlated with experimental data.
Our take: The inhibitor-bound DHX9 structure (PDB: 9MFT) was deposited in May 2025, and no other structures were available before June 2023. Boltz-2 failed to predict the correct fold for part of DHX9 (likely due to a lack of similar examples in its training data). Despite this global misfolding, the model did occasionally place ATX-968 near the known binding pocket on DHX9—which is encouraging. Its affinity predictions for ATX-968 analogues aligned with actual SAR, another surprising example in which Boltz-2’s affinity predictions remained accurate despite imprecise poses. (Note: Three of the five data points largely drive the correlation, whereas the two outliers - both predicted near 1.0 - cover the full activity range).
We also explored whether adding pocket constraints could help Boltz-2 predict the correct holo-conformation of the DHX9–ATX-968 complex. Applying these constraints recapitulated some key interactions: Boltz-2 correctly placed the ligand’s chlorobenzene (halogen) group in the pocket (crucial for the molecule’s residence time), and it positioned the ligand’s methyl-sulphonamide group in roughly the right location (though the orientation was off). Notably, that methyl-sulphonamide moiety is key for the pocket-closing interactions with DHX9’s disordered N-terminal domain. In short, providing a bit of structural guidance helped Boltz-2 recover some important features of this complex.
Key Takeaways
Our evaluation of Boltz-2 on real therapeutic targets highlighted both strengths and limitations. Here are the key takeaways from this initial deep dive:
Strengths
- Performs well on stable and familiar targets: Boltz-2’s 3D predictions generally excel on stable, rigid proteins, often capturing key interactions (for example, the binding pocket geometry in KRAS and Mpro). As expected, Boltz-2 delivers its highest accuracy on targets and ligands that resemble or are already included in its training set (for instance, a known PDB structure or a ChEMBL entry), such as for KRAS G12D and the WRN Helicase
- Reflects known SAR trends: A major benefit of Boltz-2 is its ability to qualitatively reflect structure–activity relationships. Even if the predicted pose isn’t perfect, its binding affinity scores tend to rank compounds in line with experimental potency.
Weaknesses
- Struggles with large conformational changes and induced-fit scenarios: Boltz-2 often has difficulty when a protein must undergo a big shape change or has multiple mobile domains with little precedent in the training data. If a protein needs to bend into a new shape to accommodate a ligand (like the allosteric changes in PI3K-α or WRN, or the dynamic binding required in cGAS), the unguided model usually fails to predict that rearrangement. These cases often require additional help—such as supplying a template of the alternate conformation or running a refinement step—to obtain the correct pose.
- Unrealistic poses: In highly dynamic or allosteric binding sites, Boltz-2 can misplace parts of the ligand or generate chemically unrealistic conformations. These pose errors can, in turn, throw off its affinity predictions. Even for rigid targets, Boltz-2 can sometimes confuse a ligand’s exact orientation when multiple binding modes are possible.
Open questions for the community:
- How well does Boltz-2 generalise? While Boltz-2’s training data curation is well described, the exact dataset remains unknown due to extensive post-processing. This makes it difficult to assess how well the model generalises—especially since much of ChEMBL and the PDB may be included. Access to the full training set would help clarify whether performance reflects true generalisation or overlap. More testing on novel compound–target pairs, or cases where only one modality (structure or affinity) was seen during training, would be informative. Notably, Boltz-2’s mixed performance on Recursion’s internal targets suggests reduced reliability in unfamiliar chemical space.
- Does Boltz-2 predict affinity in 3D? The model uses embeddings representing pose distributions rather than individual structures, which may enable it to learn correlations across similar ligands and targets. This raises the possibility that Boltz-2 behaves more like a QSAR or chemoproteomic model, leveraging learned associations rather than reasoning explicitly over 3D structure. Distinguishing between these two possibilities would help clarify when and why its predictions can be trusted, and we’d love to see more community efforts focused on stress-testing Boltz-2 on novel targets and probing its internal representations.
- How much can customisations improve Boltz-2 performance? Boltz-2 has advanced customization features: users can guide the model by specifying certain protein–ligand contacts (forcing it to consider a particular binding pocket), or by providing a template structure of a similar complex. In this work, however, we deliberately mostly used Boltz-2 “out of the box” without adding extra guidance. It’s likely that performance on more challenging targets could be improved by using such constraints or templates, such as in our DHX9 example.
Conclusions
Boltz-2 represents a powerful and efficient new approach for structure-based drug design. It’s particularly well-suited for targets that are rigid or already well-characterized structurally – in those cases, it can quickly generate accurate protein–ligand complexes and provide a readout of relative binding affinities, which is incredibly useful for designing and prioritizing drug candidates. On the flip side, for proteins that are “shapeshifters” or not well characterized, Boltz-2 alone may not be enough. In such situations, pairing Boltz-2 with downstream refinement methods (like molecular dynamics simulations or flexible docking), or providing it with some structural hints, can help bridge the gap. For example, explicitly including known contacts or adding constraints (such as specific residue–residue interactions or ligand-binding contacts) during the prediction could guide the model and significantly improve its performance on especially flexible or allosteric targets. In several of our test cases (e.g., KRAS and DHX9), analogues known to be more potent were indeed assigned higher scores by Boltz-2. If that level of affinity-prediction accuracy proves robust, Boltz-2 could be highly practical: it would help teams prioritise compounds for challenging targets, leaving more detailed methods to refine the exact binding modes later. However, we still do not know whether this accuracy stems mainly from overlap with the training data or from its ability to reproduce the full 3-D protein-ligand interaction landscape.
As with any new tool, these early internal tests are just a starting point. A thorough, community-driven evaluation is still needed to determine how closely targets must resemble the training set for models like Boltz-2 to yield reliable predictions. By understanding where Boltz-2 excels and where it needs support, we can use it more effectively to accelerate drug discovery. Active development is already under way to address current limitations - for example, training on more conformationally diverse data so the model can cope with proteins that adopt multiple shapes, and refining its geometry engine to better capture ligand flexibility and protein motion. These enhancements should make future versions more capable of handling allosteric sites and multi-domain targets. As with most computational tools, interpreting Boltz-2's outputs requires scientific judgment and contextual understanding. For now, Boltz-2 lowers the barrier to insights that once required resource-intensive physics-based simulations. It will provide its greatest value when teams combine their domain expertise and intuition with this fast, integrated AI tool as part of an end-to-end co-ideation platform for drug discovery.
Acknowledgments
We thank the deepmirror team for their contributions to this work. In particular, we are grateful to Tushar Modi for running the majority of the experiments, Andrea Dimitracopoulos for extensive editorial support, and Janosch Menke and Daniel Crusius for their valuable input on the writing and statistical analysis. We also appreciate the members of the broader community who provided early feedback and helped shape this evaluation including Michelle Southey, Patrick Lee, and Pat Walters.
References

References

.svg)
