.svg)

6x faster discovery with deepmirror
Optimising ADMET properties is an essential step in drug discovery to ensure new drugs are absorbed, reach the right place, are non-toxic and remain in the body the right amount of time. In this case study, we show how a biotech used DeepMirror to identify new highly potent molecular scaffolds in 2 weeks instead of 3 months.
Adverse ADMET is a common cause of drug program failure
The fast-paced world of drug discovery is full of dead ends and abandoned projects due to compounds having adverse absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. In fact, 40-45% of all drug programmes fail due to adverse ADMET (Sun et al., 2022). Once a lead series (a collection of compounds with promising binding properties) is abandoned, it is often difficult to go back and find new scaffolds that might work against a target without starting from scratch.
Recently, a European biotech that focuses on delivering first-in-class drug candidates for oncology, struggled with adverse ADMET properties on their lead series. Instead of axing the programme and starting anew they used DeepMirror to predict better lead scaffolds (a common core structure) and found them. This case study highlights how DeepMirror can be leveraged to reduce the most common failures during drug development programmes such as poor ADMET. In this case study, we detail how this was done.
Stuck with novel hit identification
For a novel unidentified target, the company used virtual screening, a method that virtually simulates how a compound binds to a pocket in the target (docking), to identify a series of promising hits (candidates). The company then underwent a “hit expansion” step, whereby they generated series of molecules like the hits. Despite successful hit expansion and achieving good potency levels for a couple of chemical series, optimizing the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties proved challenging. Further virtual screening on internal and commercial libraries failed to identify additional novel hits, prompting the need for an innovative approach (Fig.1).
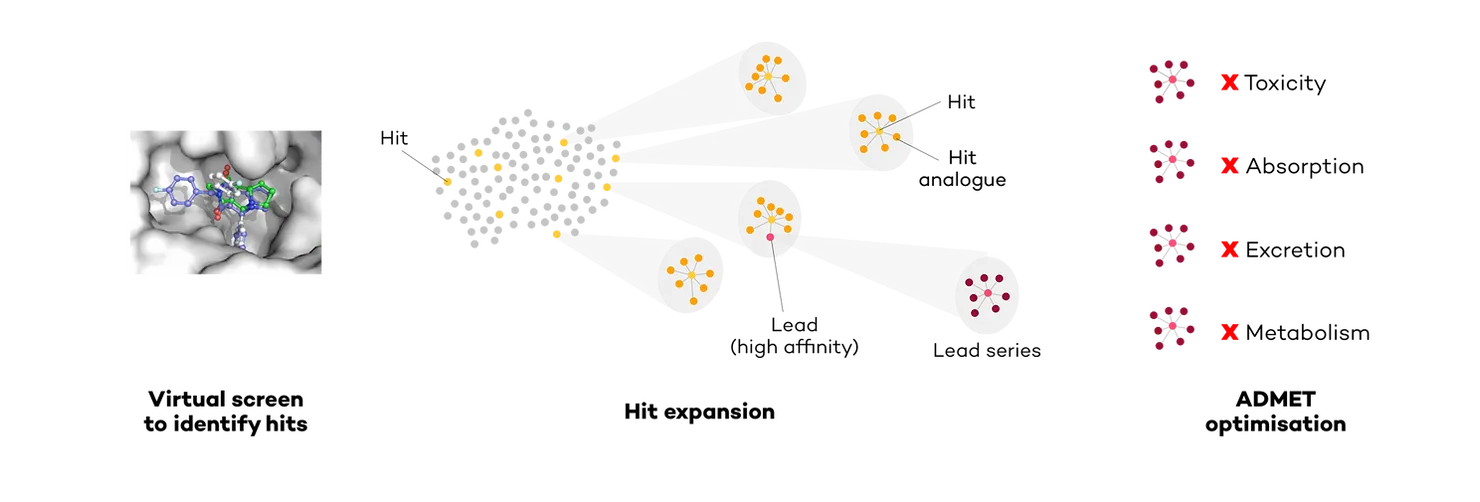
DeepMirror to find new scaffolds
To solve their ADMET issues, the company started looking into AI approaches to identify alternative lead compounds with better ADMET. AI software predicts compound activity from experimental data and can thus be more precise than docking algorithms that may struggle with complex binding sites. However, it is often technically complex and time consuming to implement AI as there are hundreds of different algorithms one can use and it is impossible to know what will work best in advance. Hence, the company decided to try out DeepMirror’s AI platform to quickly screen thousands of compounds using the data they generated so far in the hope of finding binders with better ADMET profiles.
The primary aim was to uncover novel active scaffolds (the core structure of a compound) within the company's internal chemical library that were significantly different from their previously unsuccessful leads so that they may exhibit better ADMET while still being active against the target.
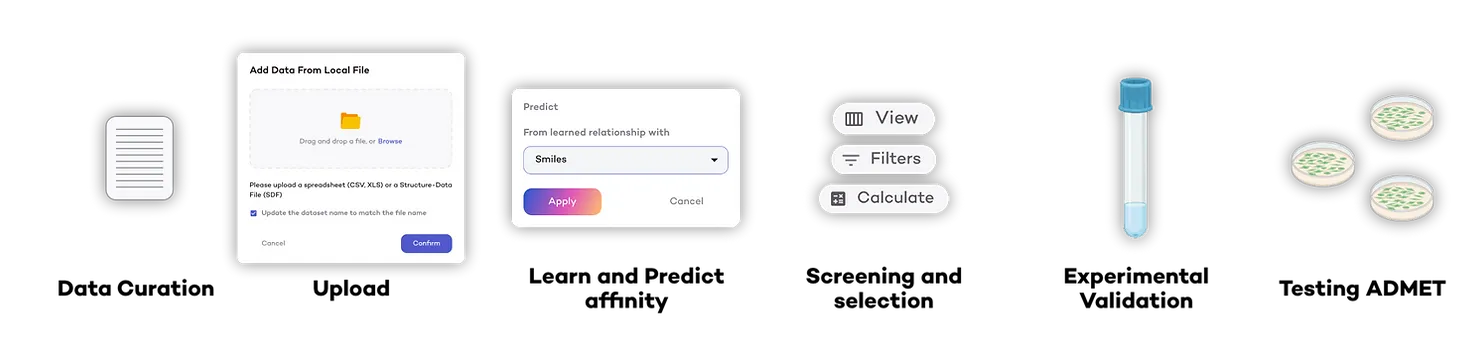
1 | Data Curation and Transformation: The team exported all compounds with measured activity data against the Target from their internal database and curated the dataset to ~400 high confidence. The data, initially in IC50 values (nM), the concentration required to inhibit the targets activity by 50%, were transformed into pIC50 (the negative log base 10 of IC50 in nM) to standardize potency measures.
2 | Uploading to DeepMirror: The curated dataset in SDF format, including structures and pIC50 values, was uploaded to DeepMirror, creating a bespoke and standardised dataset for this project.
3 | Learning affinity: DeepMirror's algorithms were employed to construct a prediction model from the uploaded pIC50 data. The model demonstrated excellent performance, with a Root Mean Square Error (RMSE) of less than 0.4, indicating high reliability in predicting compound activities.
4 | Predicting affinity, Screening and Selection: The entire internal chemical library, comprising approximately 5,000 compounds with unknown affinity against the Target was uploaded to DeepMirror. The company ranked these compounds based on their predicted activity, and the top 30 compounds were selected, consciously avoiding those with high similarity to previously studied scaffolds.
5 | Experimental Validation: Primary biological screening validated the activity of ten compounds. Further testing on chemical analogues (similar compounds to the hits) of the two most active compounds (pIC50 > 6) led to the identification of two novel scaffolds with significant biological activity against the Target. One of them was selected for further ADMET profiling.
6 | Predicting/testing ADMET: The company’s internal drug discovery platform further characterized the ADMET profile of the newly discovered chemical series. The most interesting derivatives showed very good metabolic stability, cellular permeability, and mid-low solubility, representing an optimal starting point for a new hit-to-lead campaign.
Conclusions and Future perspectives
The use of DeepMirror in this project marked a significant advancement for the company’s discovery endeavours and saved the company from testing all 5000 compounds in their library. The company estimates that by integrating proprietary internal data with DeepMirror's predictive analytics, the team overcame difficult ADMET problems, identified new active compounds and expanded their chemical arsenal against the Target with two novel scaffolds 2-3 months faster than without DeepMirror. This success story exemplifies the power of harnessing small internal datasets and state-of-the-art AI to expedite and refine the drug discovery process. The company is now working on the novel scaffolds and plans to move them into preclinical studies once optimised.
About DeepMirror
DeepMirror is the most intuitive AI drug design app that empowers chemists by predicting drug properties such as potency and ADMET so that they only synthesise the compounds that matter. The app will accelerate drug discovery by 2x, can be used by everyone within minutes, and is already deployed in biotechs across the world.

References
Sun, D., Gao, W., Hu, H., Zhou, S., 2022. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B 12, 3049. https://doi.org/10.1016/J.APSB.2022.02.002

.avif)
.svg)
