.svg)

What’s going on with AI in Drug Discovery?
Artificial intelligence is all over the headlines and promises to revolutionize drug development. There are claims that AI is "taking over drug discovery" or that it could reverse Eroom’s Law (the troubling trend of rising costs and timelines in developing new drugs). Companies like Nvidia, with significant investments in AI, are positioning it as a powerful solution for reducing costs and transforming pharma.
Beyond the hype, there is also growing skepticism. After years of anticipation, some AI-driven drug candidates have faced setbacks in clinical trials, and voices in the industry are beginning to call for a reality check.
So, is AI in drug discovery a game-changer, a letdown, or something in between? In this post, we’ll go back to the basics of AI in drug discovery, set realistic expectations, and explore the future possibilities.
What is AI?
At its core, AI uses data and algorithms to learn patterns and rules in the real world. For instance, consider using data on whether small molecules elicited toxic responses in mice as an input and output to an AI model. A model built on this data will, with enough data, learn which molecules might have harmful effects for mice. Building these models relies on data such as paired inputs (X values), like molecular structures, and outputs (Y values), like observed biological effects, which train the algorithms.
As more data is fed into the model, it uncovers patterns that help predict outcomes for new compounds resembling those in the original dataset. Think of AI as a tool that can interpolate and predict “in-between-the-dots”, enabling exploration of how slight modifications to a molecule, such as changing an R group or small scaffold changes, might influence its properties.
AI in drug discovery is not an entirely new concept. For decades, scientists have used molecular structure to predict outcomes like ADMET properties, affinity, and toxicity. In 1962, Hansch et al. introduced the concept of quantitative structure-activity relationships (QSAR), which connects a molecule’s structure to its biological effects in an animal or assay.
What has changed in recent years is the sheer scale of data that AI models can now process. Advances in deep learning allow modern AI models to handle vastly larger datasets than the few hundred molecules that traditional QSAR models once relied on. This growth has been accompanied by the rapid development of new deep learning architectures, which have driven excitement around tools like ChatGPT and similar innovations. But, as we will see below, these methods have not quite had the promised impact in Drug Discovery yet.
No (relevant) data means no AI.
It’s crucial to understand that without data, AI cannot operate. For example, if only small molecule toxicity data from mouse models is available, that data cannot be used to predict toxicity for macrocycles (see graph below). This limitation was notably demonstrated in the recent BELKA challenge, where a dataset containing triazine-based molecules with measured on-target activity was used to predict the activity of molecules without triazine. In the end, no model could successfully make this prediction (see the results).
In technical terms, this highlights AI’s strength in interpolation—predicting “in-between-the-dots”—but its weakness in extrapolation, or predicting in unfamiliar territory. This is a key limitation, as drug discovery often involves finding entirely new, previously unknown compounds. Without relevant data, AI has no basis for interpolation, and thus no ability to inform discovery.
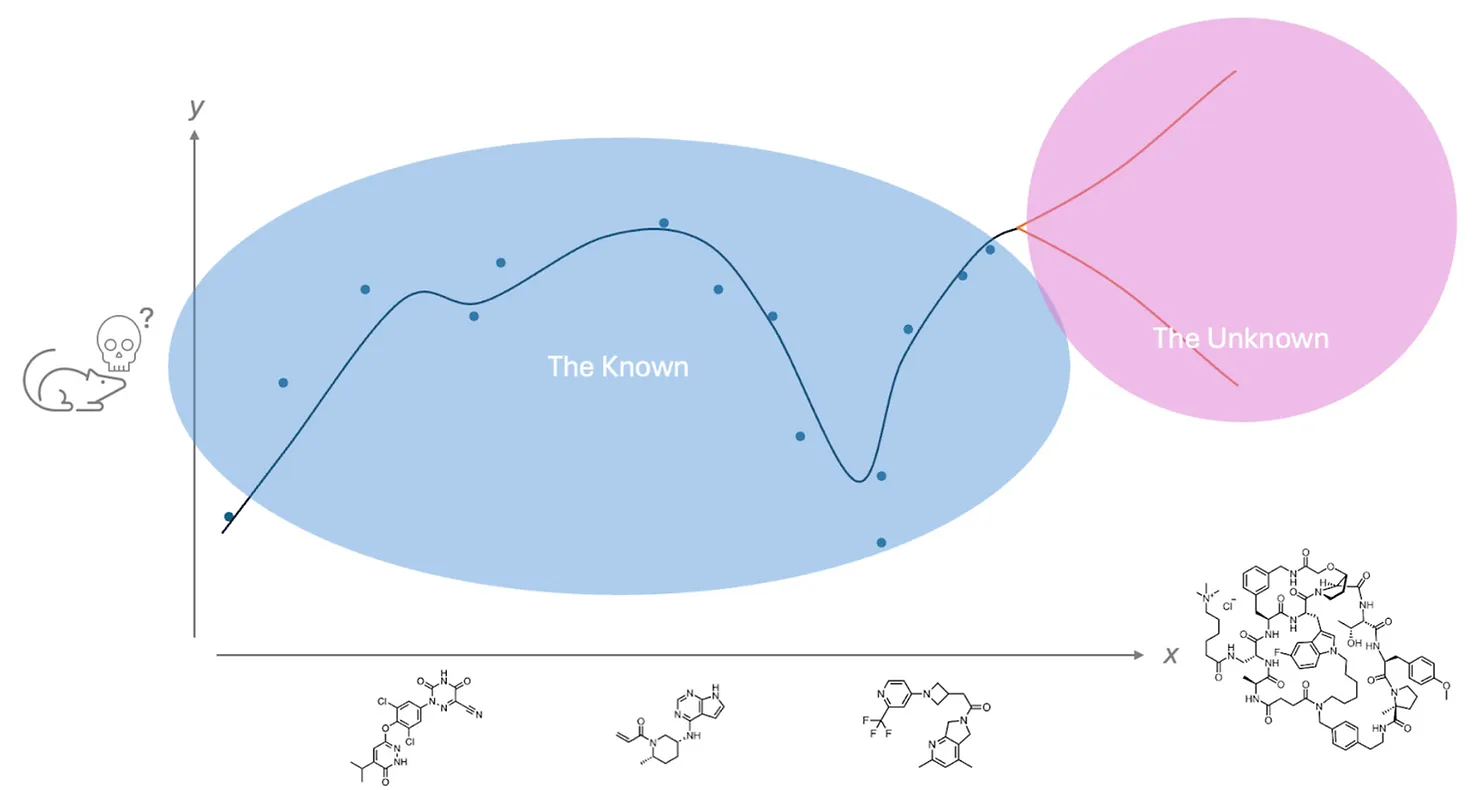
Beyond data relevance, volume is also a significant challenge. In drug discovery, available data is far less abundant than in many other fields. For example, models like ChatGPT are trained on immense datasets (around 10 trillion words) while AlphaFold was trained on approximately 200,000 protein structures. In chemistry and clinical research, data is even more limited: there are only about a thousand FDA-approved drugs, and even the largest pharmaceutical companies might have around 100,000 measured molecular properties for a single in vitro endpoint, with much less for clinically relevant ones.
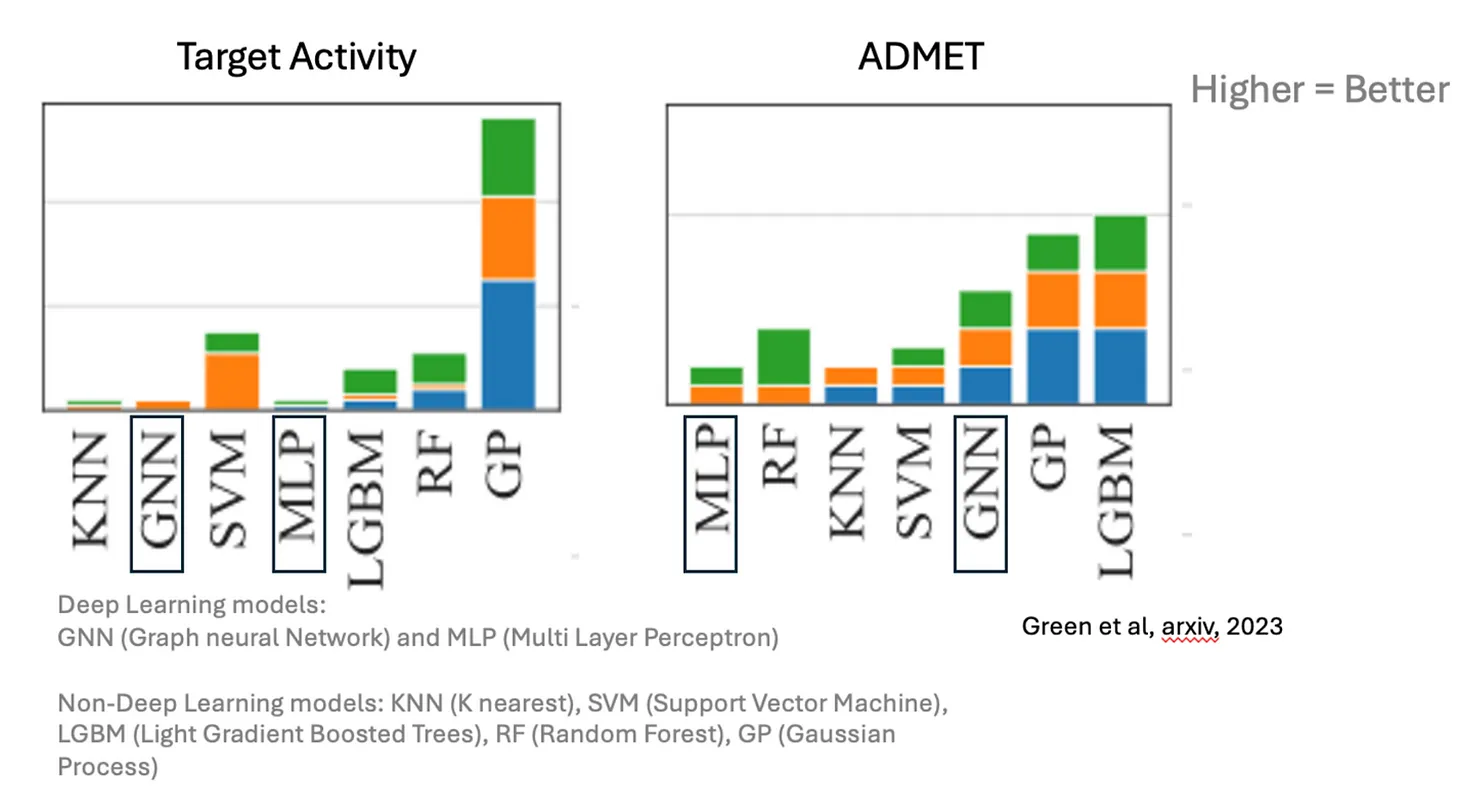
This data scarcity means that drug discovery has yet to fully benefit from the breakthroughs deep learning has enabled in other fields. AI models like graph neural networks (GNNs) and multi-layer perceptrons (MLPs) tend to improve with larger datasets, which has driven progress in areas like image and language processing. However, in predicting small molecule properties, it remains difficult to show that these newer architectures significantly outperform traditional methods.
Last year, at deepmirror, we conducted a study comparing various modelling approaches and found that while these models performed moderately well across multiple datasets, they often lagged behind classical approaches like Gaussian Processes when applied to real-world data (see plot below and our previous post).
The Reality Check
Due to the limitations in both the relevance and size of available datasets, AI's impact in drug discovery remains constrained to closely iterating over structurally related molecules. As of today, we can somewhat reliably use AI to figure out, “If I substitute an amide with a sulfonamide, what would be the effect on this assay, given the data I have?”. While our current approaches with AI have already made meaningful contributions to drug discovery programs, as we’ll discuss in an upcoming blog post, it doesn’t mean that AI can deliver a clinical candidate in just a few days. To truly transform drug development, we need to prioritize the collection and curation of extensive, relevant datasets that enable reliable predictions across uncharted chemical spaces.
AI in drug discovery is a powerful tool, but it’s not a one-size-fits-all solution. The most promising future lies in using AI to augment—not replace—traditional research methods. Success will come to those who seamlessly combine human expertise, relevant training data, and well-designed AI models with clearly defined boundaries of applicability. At DeepMirror, we are making this vision a reality by empowering chemists with intuitive AI tools that require no prior experience. Our platform eliminates the complexity of managing data and models, enabling chemists to focus on their core creative strengths while leveraging AI as a collaborative assistant.
If you’re interested in discussing these ideas further or exploring ways we might work together, feel free to get in touch!


.avif)


.svg)
