.svg)

Toward Chemical Interpretability of AI-Predicted Poses
Cofolding models like AlphaFold3, OpenFold3, Boltz-2, and Chai-1 can generate protein–ligand complexes that look convincing but often break basic chemical rules. In this post, we describe how post-processing steps such as restoring bond types, hydrogens, and realistic geometry can make these AI-predicted poses chemically interpretable and ready for downstream modeling.
Cofolding algorithms like AlphaFold3, OpenFold3, Boltz-2, and Chai-1 can predict how a small molecule (or ligand) interacts with a protein complex, e.g. to create hypotheses about a protein’s potential binding site, recapitulating experimental affinity data, and overall offering powerful structure-based drug design capabilities independently of available crystal structures.
However, cofolding models are fundamentally data-driven, they learn statistical relationships rather than obeying physical laws. They don’t account for force fields, bond energies, or ideal geometries. In our previous blog, we discovered that while these models work very well with placing ligands in well-defined and existing pockets (orthosteric), the fine structural details they generate aren’t always chemically or physically realistic.
Because of this, structures generated by cofolding should be refined before use in down stream simulations. The accuracy of free energy perturbation (FEP) calculations, for example, depends on the quality of the starting complex [1], and even small structural errors can lead to incorrect conclusions about biological activity [2]. PoseBusters can be used to assess the physical realism of molecules within protein pockets [3] and show that pose refinement improves the plausibility of ML-predicted complexes. More recently, Skrinjar et al. [4] found that as predictions deviate from training data, PoseBusters success rates drop, increasing the likelihood of unphysical poses. Together, these findings highlight that ensuring physically realistic structures through systematic pose refinement is essential when using cofolding models for FEP or other computational methods in silico.
In this post, we’ll take a closer look at some of the limitations we’ve observed when using models like Boltz-2 and Chai-1 to generate small-molecule-protein complexes, and what improvements can make their outputs far more reliable and inspire new structure–activity relationship (SAR) hypotheses that might not have been obvious before.
From our experience, two recurring problems have the biggest impact on downstream structural analysis and SAR interpretation:
- Incomplete molecular topology, leading to incorrect bond orders, missing hydrogens, or implausible atom type.
- Unrealistic ligand poses, where the molecule fails to form the expected interactions within the pocket.
These issues often arise from how the models represent chemistry internally. In other words, they are not random mistakes, but symptoms of how co-folding models simplify molecular structure. Understanding why these artifacts appear makes it much easier to fix them. In practice, we’ve consistently observed three key issues that have the greatest downstream impact:
- Missing explicit hydrogens: Many models skip hydrogen atoms entirely. Without them, it’s impossible to assign protonation states or identify hydrogen-bond donors and acceptors, both critical for interpreting binding.
- Missing bond-type information: When bond orders aren’t specified, aromatic rings, aliphatic systems, and hybridization states (sp² vs. sp³) can all be misrepresented. This can confuse chemists whose SAR hypotheses depend on accurate ring types or conjugation patterns, especially if the model outputs incorrect bond details.
- Incorrect ligand geometry: Bond lengths, angles, or torsions may be distorted, introducing unrealistic strain or breaking planarity in aromatic systems. These distortions can mislead downstream modeling or docking workflows.
These artifacts can be corrected through straightforward post-processing that restores realistic molecular geometry and completes missing chemical information. The goal is not to make predictions flawless, but to ensure they are chemically interpretable and physically consistent enough for downstream use such as docking, molecular dynamics, or free energy perturbation (FEP) calculations, where the quality of the starting structure strongly influences the outcome.
Below, we describe a series of steps to correct the poses from cofolding models. The first stage focuses on improving local ligand poses, using straightforward correction and minimal-energy optimization procedures that fix missing chemistry and relax strained geometries. The second stage goes further, refining the full protein–ligand complex by constructing force-field parameters and running explicit-solvent simulations to restore physically meaningful interactions between the ligand and its surrounding residues.
Improving local ligand poses
Step 1: Predict the ligand’s protonation state
Getting the protonation state right helps you assign the correct charges to heteroatoms and understand how the molecule actually binds, for instance, whether an oxygen is acting as a hydrogen bond donor or an acceptor. This step is crucial for mapping hydrogen-bond networks and for setting up any downstream force-field calculations. We begin by assigning the protonation state of the small-molecule SMILES using heuristic rule-based tools such as dimorphite-dl [5]. These methods rely on predefined SMARTS pattern libraries that encode atom-type environments and functional groups known to undergo protonation or deprotonation under physiological conditions. Although this strategy is inherently limited by the breadth and chemical generality of the underlying SMARTS patterns, such that uncommon, highly conjugated, or context-dependent functional groups may be misaligned, it remains computationally efficient and easily extensible. Additional SMARTS patterns can be incorporated as new chemotypes are encountered. For chemically ambiguous or electronically complex cases, quantum-mechanical, deep learning, or hybrid approaches can be used to predict pKa and assign protonation states with higher accuracy, albeit at substantially increased computational cost [6].
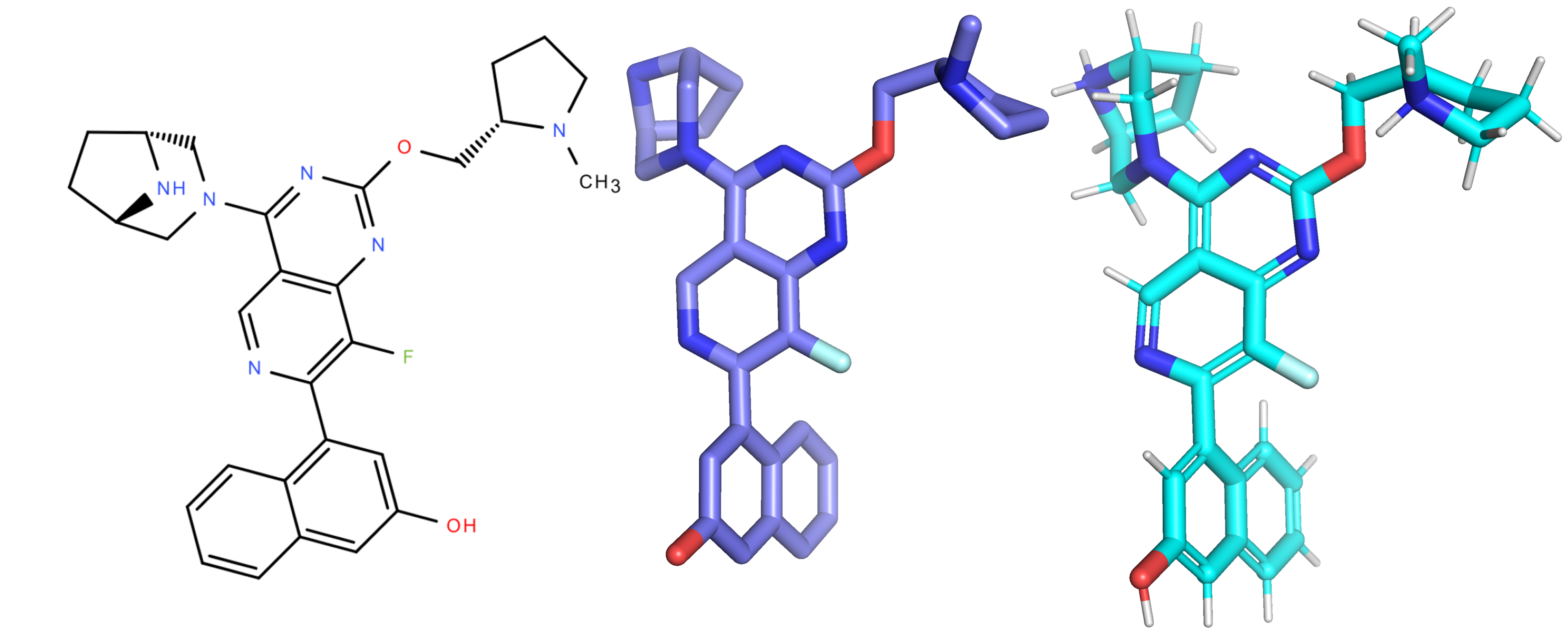
Step 2: Assign bond types and add explicit hydrogens
Once the protonation state is set, the next step is to reconstruct the original molecular topology by mapping the bond orders from the input SMILES onto the predicted 3D structure. The protonated SMILES provided for structure prediction contains all necessary bond type information (single, double, aromatic, etc.), which must be reassigned to the corresponding atom pairs in the cofolded 3D ligand. This mapping ensures that each bond in the predicted structure gets assigned the correct chemical type. With accurate bond types established, realistic geometric features can then be recovered, such as enforcing planarity in aromatic systems, distinguishing sp² from sp³ centers, and restoring appropriate hybridization patterns. See [7] and [8] for more methods on assigning bond orders.
Then, using the charge information from Step 1, adding explicit hydrogens, where they belong, ties all protein-ligand and intra-ligand interactions together. With the hydrogens in place and the correct bonds assigned, you can clearly see how the ligand interacts with the protein, whether through hydrogen bonds, π–π stacking, or other noncovalent interactions (see Figure 1).
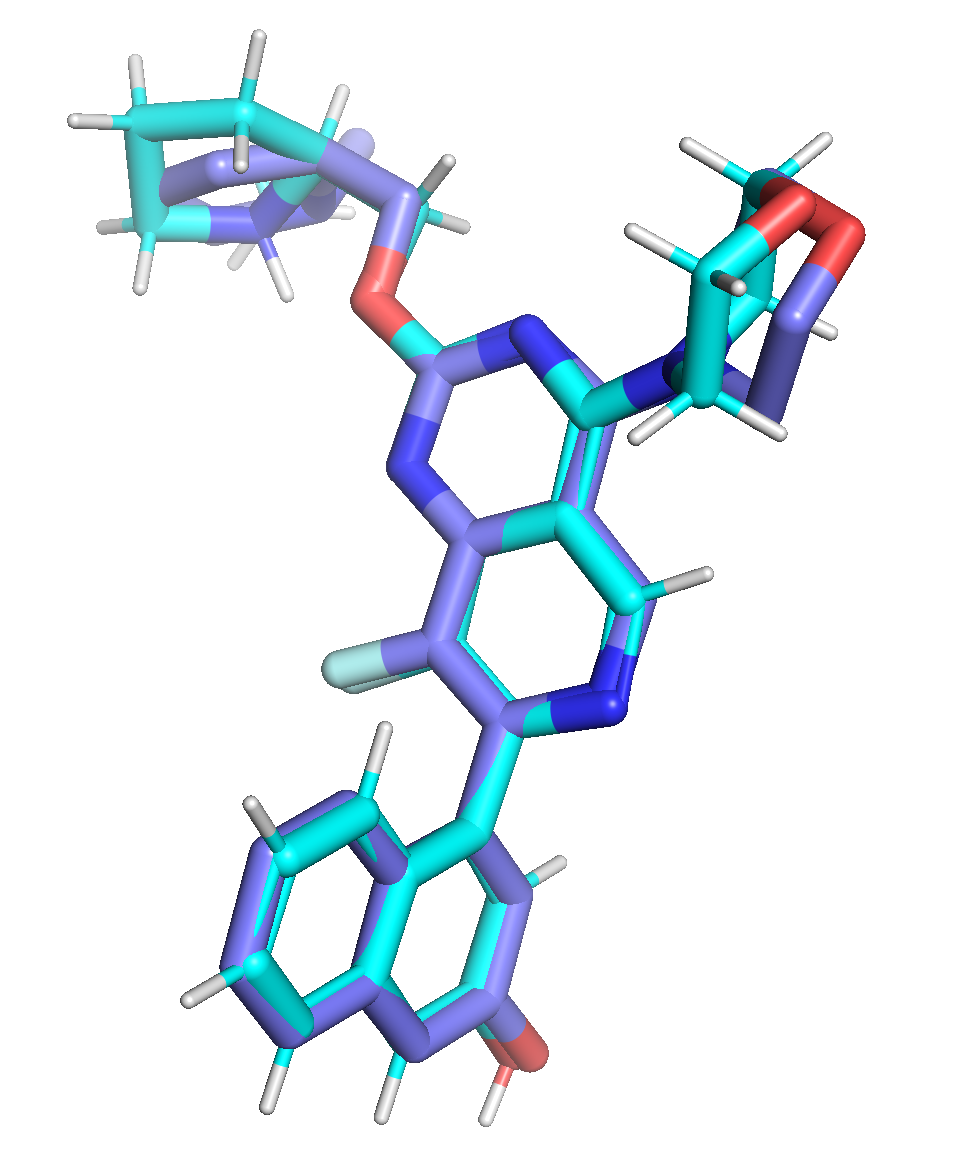
Step 3: Geometry optimization of the ligand (minimally restrained)
After fixing protonation, bond types, and hydrogens, the ligand’s geometry may still be slightly off, with non-ideal bond lengths, strained angles, or incorrect torsions. To clean this up, we run a short geometry optimization on the predicted ligand structure to gently relax it within the binding site. In order to preserve the overall pose predicted by the model, we apply harmonic restraints to the heavy atoms. This keeps the ligand anchored in roughly the same position while allowing just enough flexibility to correct local distortions in bond lengths, angles, and torsions [9].
For this step, we use the MMFF94 force field in the gas phase (i.e., without explicit solvent), which is well-suited for small-molecule geometry refinement [10]. This approach removes minor structural flaws at a modest computational cost, producing chemically sensible conformations without drifting far from the predicted pose.
Refining interactions with the protein environment
Even after local cleanup of the ligand, the surrounding protein environment can show steric overlaps or unrealistic side-chain orientations around the ligand, leading to van der Waals clashes and poor local geometry. These artifacts happen because cofolding models optimize for global structural plausibility, not for detailed atomic interactions. As a result, what looks stable overall may still be chemically strained or physically impossible at the atomistic level.
Such distortions can undermine the confidence in the predicted pose and make it less useful for downstream modelling or quantitative analysis. In fact, recent benchmarking studies have shown that deep-learning cofolding models often generate non-physical structures with steric clashes or stretched bonds [2]. To fix this, we can perform a full refinement of the protein–ligand complex under explicit solvation, restoring realistic packing and hydrogen-bonding interactions.
Once the ligand and its local geometry are corrected, the next step is to ensure that the entire protein-ligand complex behaves realistically. This involves resolving steric clashes between the ligand and nearby residues, optimizing side-chain conformations, and accounting for solvent effects, which are often crucial for accurate binding interactions.
In practice, we carry out a full refinement of the cofolded complex under explicit solvation. This process relaxes the structure producing a model that’s ready for downstream analysis.
The workflow consists of several key steps:
- Ligand parameterization: The protonated ligand (with explicit hydrogens and charges) is parameterized using the GAFF2 force field for small molecules, which employs QM-corrected atomic charge distributions (using correction of AM1-BCC charges [12], used for the KRAS example discussed in the blog) [13]. Any other cofactors present in the cofolded structure are treated in the same way (see tutorial [14])
- Assigning force fields to biomolecular components: To ensure consistency across the system, the following force fields are applied (see tutorial [15]):
- Protein: AMBER ff14SB [16].
- DNA / RNA: DNA.OL24 / RNA.OL3 [17-18].
- Ions: 12-6-4 LJ-type nonbonded model that accounts for polarization effects [19].
- Solvation: The assembled complex is then solvated using the TIP3P water model, which captures realistic hydrogen-bond networks and solvent-driven interactions [20].
- Multi-stage energy minimization (see tutorial [21])
Finally, the system is gradually relaxed in stages to remove local strain without distorting the predicted binding pose:
- Minimize the solvent only
- Minimize solvent + ligand
- Minimize all atoms together
This stepwise relaxation efficiently resolves steric clashes and rebalances local geometry while preserving the overall cofolded arrangement (see Figure 3). It is worth noting that other platforms are actively adopting similar pose-refinement/ post-processing pipeline which help alleviate some of the limitations of the inaccuracies generated by cofolding predictions [1].

Conclusions
Cofolding models such as AlphaFold3, OpenFold3, Boltz-2, and Chai-1 offer promising new ways to explore protein–ligand interactions, but their predictions should be interpreted with caution. While these models often capture general binding modes well, they can overlook the detailed chemistry required for physically realistic poses. By complementing deep-learning predictions with post-processing steps that restore protonation states, bond orders, and local geometry, it becomes possible to close the gap between statistical inference and chemical structure. This refinement improves the physical plausibility of predicted complexes, making them more suitable as starting points for downstream computational chemistry workflows such as docking, free energy perturbation (FEP), or molecular dynamics simulations.
Acknowledgments
We thank the deepmirror team and our scientific advisors for their contributions to this work. In particular, we are grateful to Tushar Modi for running the majority of the experiments and writing.
Reference
[1] https://arxiv.org/html/2508.19385v1
[2] https://www.nature.com/articles/s41467-025-63947-5
[3] https://pubs.rsc.org/en/content/articlelanding/2024/sc/d3sc04185a
[4] https://www.biorxiv.org/content/10.1101/2025.02.03.636309v3.full.pdf
[5] https://github.com/durrantlab/dimorphite_dl
[6] https://link.springer.com/article/10.1007/s10822-020-00362-6
[8] https://practicalcheminformatics.blogspot.com/2018/09/assigning-bond-orders-to-pdb-ligands.html
[11] https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01688
[12] https://pubmed.ncbi.nlm.nih.gov/12395429/
[13] https://doi.org/10.1021/acs.jctc.8b01039
[14] https://ambermd.org/tutorials/basic/tutorial4b/index.php
[15] https://ambermd.org/tutorials/basic/tutorial7/index.php
[16] https://doi.org/10.1021/acs.jctc.5b00255
[17] https://pubs.acs.org/doi/10.1021/acs.jctc.4c01100
[18] https://pubs.acs.org/doi/10.1021/ct100481h
[19] https://pubs.acs.org/doi/10.1021/jp505875v
[20] https://doi.org/10.1063/1.445869
[21] https://docs.openmm.org/latest/userguide/application/02_running_sims.html
-




.svg)
